![]()
![]()
![]()
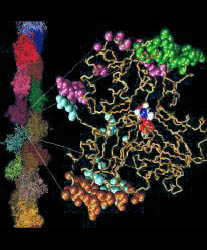
The Tcl (John Ousterhout, 1988) version of Hingefind is designed for interactive use with the free molecular graphics package VMD. This version does not maintain the spatial connectivity of the found domains, as implemented in the X-PLOR version. The VMD version has, however, the advantage that it only requires PDB files as input. Thereby, selections of atoms are easier to adapt to the user's needs than with X-PLOR. E.g., conformations of nonidentical proteins can be compared, as long as pairs of atoms in both structures can be matched sequentially.
![]()
There is a single file in this directory which contains the algorithm:
source hingefind.tcl
in the VMD command shell and return. The default setup of the script loads two structures of lactoferrin from the PDB databank. To load these structures, type
load
and return. The procedure load now initializes the program, selects the pairs of corresponding atoms (default: alpha carbons) and initializes some global variables. To partition the protein structures with a given tolerance epsilon, type
partition epsilon
and return. Note that the tolerance epsilon must be entered in Angstrom units (this is different from the X-PLOR implementation). The tolerance should be larger than the imprecision of the coordinates in the two structures (local noise level), but smaller than the total rms deviation of the two conformations. The protein now iteratively finds the rigid domains, sorts them by size, and draws tubes in the color of the domain-id, starting at domain-id 0, the largest found domain (note that this is also different from the X-PLOR version, where domains are numbered starting at 1). To determine the effective rotation axis (hinge) and other information about the relative movement of two domains domid1 and domid2, enter
hinge domid1 domid2
and return. Domain domid1 is the reference domain and domain domid2 the moving domain. Both structures are superimposed by the reference domain. The rotation axis is shown as an arrow with it's orientation representing a left-handed rotation about the axis. A "pivot" point can be found in the middle of the arrow which is connected to the COM of the moving domain in both structures to illustrate the rotation angle. The rotation angle and other useful information about the run, the domains, and the accuracy of the rotational fitting can be found in the output (VMD command shell).
An example VMD session would look like this:
source hingefind.tcl
load
partition 2.0
hinge 0 1
hinge 0 2
partition 1.8
hinge 0 1
The initial rendering of the loaded molecules may also be altered in procedure load. By default molecule 1 is shown in purple and codes with colored tubes for the found domains, while molecule 2 remains a solid white line which follows the backbone.
![]()
The rotation angle and other useful information about the hinge rotation, the domains, and the accuracy of the rotational fitting are written to the VMD command shell. The information includes:
![]()
A programmer can find more information about available VMD commands and routines at the VMD user's guide. All procedures and variables are documented in the program. The datastructures used to store structural information and domain membership are briefly described here:
![]()
![]()