Hingefind (X-PLOR) -
an algorithm to investigate domain motions in proteins.
9-24-96
This page contains documentation specifically for the X-PLOR version.
Content:

The X-PLOR
(Axel T. Brunger, 1992) version is designed for longer batch jobs which
allow the user to run a large number of trial runs in various modes.
The X-PLOR version has all the features described in the paper. In
particular, it allows to maintain the spatial connectivity of the found
domains. The version requires that the user obtains a free license for the X-PLOR software. The output psf and pdb files can be
visualized with any standard graphics package.
There are several files and scripts in this directory to set up the algorithm:
hingefind
- A unix shell script that runs the X-PLOR job and writes three
X-PLOR stream files which contain commands from which X-PLOR
can compute filenames and the tolerance used the algorithm. It is
recommended to run the cases within a range of tolerances
between 50 and 100% of the initial rms deviation.
There are case descriptors from which hingefind.inp computes the
actual coordinate files (in pdb format) of the structures used in the
comparison, i.e. here: foo.COO, soo.COO, bar.COO. Change these to your
own filenames. Note that hingefind.inp will always interpret "foobar"
as "foobar.COO", so you may want to edit hingefind.inp if you use other
file suffixes (e.g. ".pdb") instead of ".COO".
partition.str
- A stream file which contains necessary X-PLOR commands to set
up the structure. It may contain a pointer to a psf file. The segid
"APO" is be used for the parts of the protein which should be aligned,
otherwise hingefind.inp has to be modified. Note that the coordinates
in the two compared pdb files must be both compatible with the
structure. The pdb files may contain additional atoms which do
not have to be specified in partition.str if not used in the
alignment. To appear in the output files, such additional structures
which are not used in the alignment should be given a segid different
from "APO" in partition.str.
dum.top
- An X-PLOR topology file with the residues of dummy molecules
used in the algorithm for visualization of pivot points and
axes.
prexplor.dim
- The X-PLOR file which contains array sizes for compilation
(35,000 atom version). It will probably be necessary to
compile X-PLOR with the larger BUFMAX parameter for the loops.
This executable is named "xl" in the hingefind script.
hingefind.inp
- The X-PLOR script with the algorithm. There are a variety of
variables and paths the user has to specify in the head of the
file :
- $ndomains: The max. number of domains to be found (<= 999).
Recommended: 999 to yield full partitioning. Small values (2-5)
should be used for test runs.
- $maxccounter: The number of maximum cycles of the
"converge" loop. In case the algorithm does not converge
within the specified number of cycles (this was very rarely
observed to occur in the "fas" partitioning mode at extreme
tolerances), a warning message is written in the log file.
Recommended: 10 - 20.
- $ptmeth: This variable determines the mode of the partitioning part
of the script: "man" specifies manual assignment of domains,
no partitioning. Up to 9 domains can be assigned and
$ndomains must be smaller than 10. "fas" codes for the fast
version of the automatic partitioning algorithm, in which the
connectivity of the residues in the found domains is NOT
maintained. "slo" specifies the slow partitioning algorithm
with maintained connectivity of the domains.
- $subset: This variable determines the subset used to partition
protein atoms with segid "APO" for $ptmeth = "slo" or $ptmeth = "fas".
"cao" specifies C-alpha atoms only;
"bac" specifies the backbone atoms with name C, CA, N;
- $cutdom: Many found domains will be very small in size. This cutoff value
for the residue number determines minimum domain size of a domain for
determination of effective rotations.
- store1...9: The selection attributes which allow the assignment of
up to 9 domains by hand in the "man" mode.
- $case1COO: String that specifies input file for the coordinates in
pdb format or pointer to pdb file. The path has to be
specified. X-PLOR can compute the filename from the variable
$case1 defined in the streamfile casefile1 written by the
shell script. Coordinates written to main coordinate set.
- $case2COO: String for 2nd pdb file (comparison coordinate set
Analogous to $case1COO.
- $oname: Output pdb file with assigned domains, pivots, axes.
The filename will be computed using some of the above variables and
the $fname variable which contains the tolerance as defined by the
shell script.
- $uname: Output psf file, analogous to $oname.
- $dname: Output log file with information about the proposed
effective rotations, residues, accuracies.
Running a particular system with a range of tolerances in "fas" mode,
it was found that there exists one or more windows of optimum tolerance
where the relative errors were very small. Therefore it is recommended
to try a range of tolerances first with the "fas" mode, find the
window(s) of small error and then calculate selected tolerances in the
window(s) in "slo" mode with a higher number of domains. The error of
the domain fitting was found to decrease with "slo" partitioning due to the connectivity of the domains.
There are three output files specified by the variables $oname $uname
and $dname: pdb and psf files of the labeled structure, and the log
file of the run. The pdb and psf files can be used to visualize the
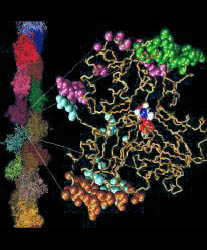
results of the algorithm: The data is labeled by segid's:
- "D0" is the unconverged rest of the protein,
- "D1" is the reference domain of the protein ("rigid core"),
- "D2", "D3", etc, are additional domains,
- "R" denotes small domains with a size below $cutdom (see 2.),
- "A2", "A3", etc, are the dummy molecules which
visualize the effective rotation of the domains.
In addition, the user may also find untouched atoms with the segid "APO" and other segid's not used in the alignment.
The
dummy molecules show an arrow along the rotation axis with it's
orientation representing the left-handed rotation about the axis. The
constructed "pivot" can be found in the middle of the arrow and is
connected to the COM of the main and comparison coordinate set of the
domain to illustrate the rotation angle. The rotation angle and other
useful information about
the run, the domains, and the accuracy of the rotational fitting can be
found in the self-explanatory log file.
NOTE: The X-PLOR log files would contain several Mbytes of data for
each run, so the standard output is piped to /dev/null. The standard
output should only be used for debugging of modified or augmented
scripts.
Other Links
 Domain Movements
Hingefind
Hingefind Tcl
Domain Movements
Hingefind
Hingefind Tcl